Czym jest histiocytoza i jak ją rozpoznać i leczyć u dzieci i dorosłych?
Histiocytoza jest bardzo rzadką chorobą układu krwiotwórczego. Już ponad 100 lat temu rozpoznano pierwsze przypadki schorzeń o nietypowym obrazie klinicznym, które z czasem zakwalifikowano do jednej grupy zaburzeń histiocytarnych. Schorzenia te polegają na tym, że złożony układ odpornościowy organizmu człowieka, zamiast go bronić, zaczyna stanowić dla niego zagrożenie. Histiocytoza stanowi dla lekarzy ogromne wyzwanie zarówno pod względem diagnostycznym jak i leczniczym.
Histiocytoza – czym jest?
Histiocytoza to choroba opisywana początkowo głównie u dzieci. Mimo, iż pierwsze przypadki wykryto kilkadziesiąt, a nawet 100 lat temu, to dopiero z czasem udało się poznać mechanizm histiocytozy. Jednocześnie dokonano podziału tej grupy schorzeń w zależności od typu komórek gromadzących się w różnych tkankach organizmu.
Kwalifikacja histiocytozy:
- histiocytoza z komórek Langerhansa;
- histiocytoza z nieokreślonych komórek (nieokreślony nowotwór z komórek dendrytycznych);
- choroba Ersheima i Chestera;
- rozsiany młodzieńczy żółtakoziarniniak;
- grupa histiocytoz z komórek innych niż Langerhansa (np. zespół niebieskich histiocytów);
- złośliwe zaburzenia histiocytarne, w tym mięsak histiocytarny, mięsak z komórek Langerhansa, mięsak z komórek dendrytycznych;
- histiocytozy złośliwe wtórne do innego nowotworu układu krwiotwórczego.
Najlepiej poznaną grupę histiocytoz stanowi histiocytoza z komórek Langerhansa i to na niej skupiona będzie dalsza część artykułu.

Histiocytoza z komórek Langerhansa – charakterystyka schorzenia
Komórki Langerhansa to komórki układu odpornościowego, które powstają w szpiku kostnym, a następnie migrują do skóry. Fizjologicznie ich zadaniem jest rozpoznawać obce antygeny, alarmować organizm o zagrożeniu i inicjować reakcje immunologiczne.
Histiocytoza z komórek Langerhansa to schorzenie polegające na rozroście nieprawidłowych komórek Langerhansa. Nacieki tych komórek mogą dotyczyć jednego lub wielu narządów. Histocytoza jest schorzeniem dominującym u dzieci, ponieważ zdecydowana większość przypadków wykrywana jest poniżej 10 roku życia. Zdecydowanie rzadziej zdarza się, by przypadki histiocytozy zostały rozpoznane u młodych dorosłych. Tak czy inaczej, rozpoznanie histiocytozy z reguły przypada poniżej 30 roku życia. Zapadalność to ok. 1-2 przypadki na milion. Zdecydowanie bardziej predysponowana jest płeć męska do wystąpienia tego schorzenia. Do tej pory nie odpowiedziano na pytanie, co jest bezpośrednią przyczyną powstania histiocytozy.
Mechanizm choroby polega na niekontrolowanym, nieprawidłowym rozroście komórek Langerhansa być może w efekcie nieprawidłowej stymulacji układu immunologicznego bądź zaburzeń tego układu. Histiocytoza z komórek Langerhansa może zająć każdy narząd, choć najczęściej obserwuje się nacieki w skórze i kościach. W dalszej kolejności obserwuje się nacieki węzłów chłonnych, płuc, wątroby, śledziony czy ośrodkowego układu nerwowego.
Histiocytozę można podzielić w zależności od zajęcia poszczególnych narządów na histiocytozę jednosystemową lub wielosystemową.
Histiocytoza jednosystemowa może być jednoogniskowa lub wieloogniskowa w obrębie określonego układu lub narządu:
- kości;
- skóra;
- węzeł chłonny;
- płuca;
- ośrodkowy układ nerwowy;
- jelita;
- grasica.
Histiocytoza wielosystemowa obejmuje histiocytozę z zajęciem przynajmniej dwóch narządów bądź układów. Wśród postaci histiocytozy wielosystemowej wyróżnia się przypadki histiocytozy z zajęciem układów krytycznych takich jak wątroba, śledziona, ośrodkowy układ nerwowy czy szpik kostny, jak również histiocytozę bez zajęcia układów krytycznych.

Jakie objawy daje histiocytoza?
Histiocytoza z komórek Langerhansa, jak już wcześniej wspomniano, może dotyczyć jednego układu bądź wielu. To też przekłada się na przebieg choroby. Schorzenie ma najczęściej kilkuletni przebieg. Konieczne jest odpowiednie leczenie, choć zdarza się, iż pojedyncze ogniska ulegają samoistnemu wycofaniu się. Schorzenie może mieć charakter nawrotowy i niestety jest to choroba o bardzo nieprzewidywalnym przebiegu. U chorych, u których obserwuje się zajęcie pojedynczego narządu, najczęściej przebieg jest łagodny. W przypadkach zajęcia kilku układów bądź narządów, a tym bardziej przy słabej odpowiedzi organizmu na stosowane leczenie, choroba ta może okazać się śmiertelna.
Początkowo objawy choroby mogą być bardzo dyskretne. Wszystko zależy od tego jakie narządy zostają objęte chorobą i jak rozległy jest ten proces. Wśród objawów ogólnych wymienia się gorączkę, nocne poty czy osłabienie. Są to więc bardzo nieswoiste objawy, choć często naprowadzające na diagnostykę procesów rozrostowych.

Objawy związane z naciekami w poszczególnych narządach:
- wytrzeszcz gałki ocznej,
- ubytki kostne,
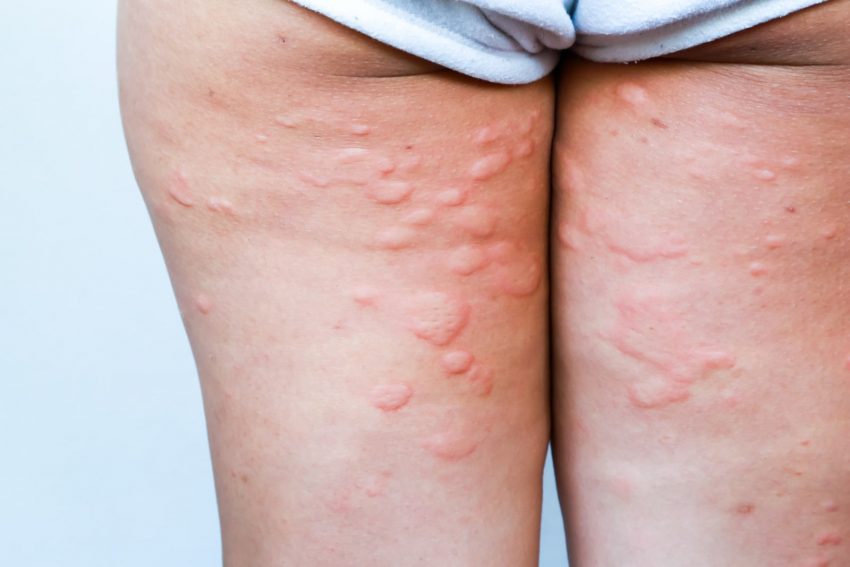
- wypryski grudkowe na skórze,
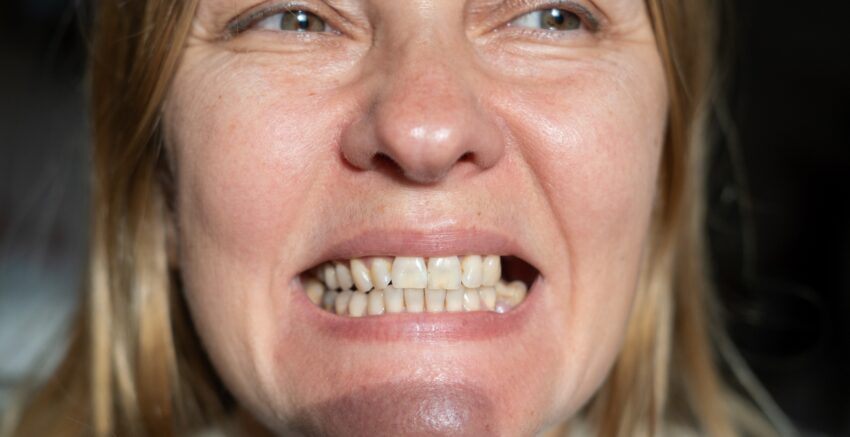
- bolesność i przerost dziąseł,
- owrzodzenia na błonach śluzowych jamy ustnej,
- patologiczna ruchomość zębów,
- zmiany łojotokowe głównie na skórze pokrywy czaszki,
- wrzodziejące zmiany w fałdach skórnych,
- patologiczny wyciek z przewodu słuchowego zewnętrznego,
- powiększenie wątroby i śledziony,
- powiększenie węzłów chłonnych,
- zmiany śródmiąższowe płuc objawiające się kaszlem i niespecyficznymi dolegliwościami bólowymi klatki piersiowej,
- zaburzenia endokrynologiczne związane z naciekiem na przysadkę mózgową,
- niespecyficzne objawy ze strony ośrodkowego układu nerwowego np. zaburzenia zachowania czy inne objawy ogniskowe.
Jak widać, objawy te nie są charakterystyczne, dlatego od momentu pojawienia się pierwszych symptomów do właściwego rozpoznania, niejednokrotnie mija kilka miesięcy czy nawet lat. To wszystko zależy od nasilenia objawów chorobowych. Jak wspomniano, najczęściej zajmowane są kości i skóra. U dzieci głównie dotyczy to kości czaszki, kręgosłupa czy żeber, natomiast u dorosłych najczęściej pierwsze zmiany są w obrębie kości żuchwy czy miednicy.
Zmiany skórne w przebiegu histiocytozy nie są specyficzne i mogą imitować wiele innych procesów chorobowych, najczęściej zakażenia grzybicze.

Jak rozpoznać histiocytozę?
Zdecydowanie łatwiej jest, kiedy histiocytoza przebiega ze zmianami w kośćcu. Są one dość specyficzne w badaniach obrazowych, dlatego już podstawowe badania tego typu mogą właściwie ukierunkować dalszą diagnostykę. W pierwszej kolejności wykonuje się szereg badań laboratoryjnych, w tym badanie morfologii krwi, badania biochemiczne, układ krzepnięcia. Jednocześnie należy wykonać diagnostykę obrazową kośćca, do czego służy badanie rentgenowskie (RTG) i tomografia komputerowa (TK). Wśród badań obrazowych wykonywanych w czasie diagnostyki znaczenie ma również USG celem oceny wątroby i śledziony, a także MR głowy pod kątem zajęcia ośrodkowego układu nerwowego czy przysadki mózgowej. Do szerszej diagnostyki obrazowej wykorzystuje się również badanie PET.
Leczenie i rokowanie w histiocytozie
Rokowanie w przypadku histiocytozy jest różne w zależności od rozległości procesu chorobowego. Jeśli naciek obejmuje pojedynczy narząd, rokowanie jest bardzo dobre. Pojedyncze ogniska czasami nie wymagają leczenia, gdyż wycofują się samoistnie. Zdecydowanie gorsze rokowanie mają procesy obejmujące szpik kostny, ośrodkowy układ nerwowy, wątrobę czy śledzionę.
Miejscowe leczenie histiocytozy opiera się na fototerapii czy stosowaniu glikokortykosteroidów. Zmiany kostne wymagają jednoczasowego leczenia ortopedycznego i niejednokrotnie radioterapii. W przypadku zmian wieloogniskowych, obejmujących kilka układów włącza się leczenie ogólnoustrojowe.
Leczenie I rzutu – leki zawierające substancje czynne: winblastyna + prednizolon ok. 12 miesięcy.
Leczenie II rzutu – prednizon +winblastyna + cytarabina przez ok. 24 miesiące. W leczeniu podtrzymującym stosuje się 6-merkaptopurynę.
W kolejnych fazach rozważa się bardziej złożone i obciążające dla organizmu leczenie farmakologiczne a ostateczności (szczególnie u dzieci) stosuje się przeszczepy allogeniczne komórek krwiotwórczych (czyli przeszczepy od dawcy zgodnego).
Źródła:
- Raciborska A, Rogowska E, Słowińska M., Pacjent z histiocytozą z komórek Langerhansa. Co ważnego dla pediatry, Pediatria Dyplom 2018,3:75-84
- Choroby wewnętrzne, Szczeklik A.(red), Medycyna Praktyczna, Kraków 2022.
Podoba Ci się ten artykuł?
Polecamy

„Torbiele najczęściej nie dają dolegliwości, ale zdarzają się i takie, które powodują bóle o charakterze rozpierającym”. Czy torbieli w piersi trzeba się bać? Tłumaczy dr Jacek Tulimowski

„Problemy hormonalne nie biorą się znikąd”. Katarzyna Wróbel o regulowaniu hormonów

Lekarka przez lata zmagała się z otyłością. „Żyłam w nieświadomości, że jestem chora”

„Uważność to potężne narzędzie w procesie wychodzenia z nałogu” – mówi terapeutka Julie S. Kraft
się ten artykuł?